Sickle Cell Disease (SCD) describes a condition in which most of the oxygen-carrying red pigment (hemoglobin) in a person’s red blood cells is type S (sickle). Under certain conditions these red blood cells become an unusual sickle shape, instead of remaining round. These cells break down more quickly than normal ones, causing anaemia (low blood count). These can also stick to each other, to other cells and to the lining of the blood vessels. This can cause blockage of blood vessels and damage to the vessel walls. It can affect all the systems in the body because they are all dependent on their blood supply.
Common complications include bone pain, Acute Chest Syndrome which is like Pneumonia and serious infections. Sickle cell disease is very unpredictable. Affected persons vary widely in which complications they get and when they occur; some people are relatively well.
Common Conditions of SCD
Acute Pain Crisis
Acute Chest Syndrome
Stroke
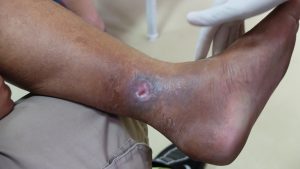
Leg Ulcers
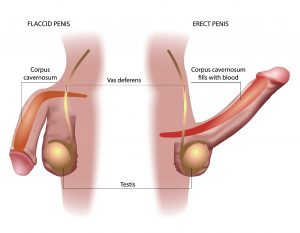
Priapism (prolonged erection of the penis)
Enuresis (Inability to control urination)
Sickle cell disease is not infectious (catching). It is always inherited from the parents. Both parents must have an abnormal gene. At least one must be the Sickle (S) gene. About 10% of the Jamaican population carry the S gene while another 5% carry other genes that are associated with SCD.
It is important for all persons to be aware of their sickle status, that of their current or potential life partner and those of their children.
In an aim to improve the management of Sickle Cell Disease, a new born screening programme has been implemented in which babies are screened immediately after birth for the disease using a dried blood sample. Once the condition is confirmed, preventative care can start as early as 6 weeks and is important as the severity of the disease is higher in early childhood. This can decrease preventable deaths.
Persons living with SCD are encouraged to practice proper management of the disease at home, avoiding triggers of pain, taking their medications when prescribed and taking extra care in food hygiene to prevent salmonella. It is also critically important that they keep their clinic visits, which will allow full immunization, and a full understanding of their disorder, including the complication common at their own age.
In keeping healthy persons living with SCD can also:
Anticipate situations that trigger a crisis such as the cold or rain;
Stay well hydrated;
Maintain a healthy diet rich in iron and inclusive of fruits and vegetables but not take iron supplements unless specifically recommended by their doctor;
If necessary, take folic acid;
Exercising as tolerated, without triggering pain.
Myths and Facts
Myth: SCD is contagious.
Fact: SCD is a genetically inherited disease. As such it is not an infection.
Myth: Only people of African descent have SCD
Fact: SCD is not a “black person” disease. While more common in African descent, it is seen to occur in other races.
Myth: People with SCD only live to their 20s.
Fact: Persons with SCD do not all die young. The average lifespan of person severely affected by SCD is 10 years less than persons without SCD. This is approximately 55 years old.
Myth: Women with SCD are infertile.
Fact: Women with SCD have normal fecundity. That is, they are able to get pregnant. It must be noted however that they have an increased risk of complications during pregnancy and delivery.
Visit the NHF website, https://www.nhf.org.jm/ , to see how you can get your Sickle Cell Disease drugs (covered under the NHF act) subsidized.